Published on 11 Nov 25
Over four decades since its conception in 1980, Fragment-Based Drug Discovery (FBDD) has emerged as an innovative and efficient approach to modern drug development, showing advantages over traditional high-throughput screening (HTS) methods. By focusing on smaller, simpler molecules, FBDD provides a foundation for developing novel therapeutics, especially for challenging previously “undruggable” targets.
Historically, FBDD has led to several milestone achievements in drug development, with eight FDA-approved drugs and more than 50 compounds currently in clinical stages. The first FDA-approved FBDD-derived drug, Zelboraf (vemurafenib, PLX4032), a BRAF inhibitor for melanoma developed by Plexxikon was initiated in 2005 and approved in 2011, demonstrating the efficiency of this approach in accelerating drug discovery timelines.
At o2h discovery, our integrated fragment-based screening platform combines biophysical and biochemical tools like the Biacore T200 Surface Plasmon Resonance (SPR) to enhance fragment screening, ensuring precision and efficiency in the FBDD workflow. This integrated approach accelerates hit identification, strengthens validation, and drives data-driven lead optimisation.
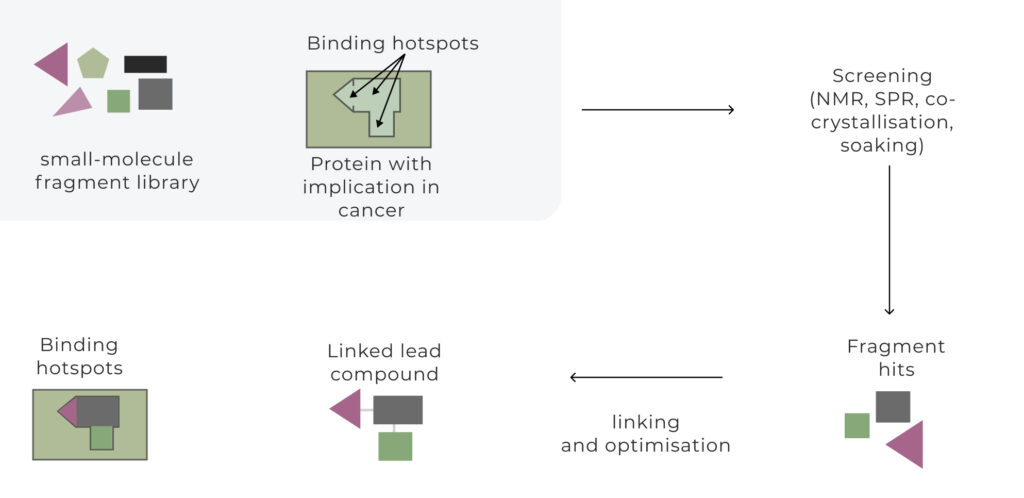
Understanding Fragment-Based Drug Discovery
Fragment-based drug discovery is a strategic approach to identifying potential start points for drug discovery projects by screening small, low-molecular-weight molecules termed fragments. These fragments bind to specific regions of a target protein, serving as a starting point for further expansion and optimization. Unlike HTS, which screens vast libraries of large, complex drug-like molecules, FBDD relies on smaller libraries of simpler compounds. This ensures better “chemical space” coverage, as the smaller size and simplicity of fragments allow for a more comprehensive exploration of potential interactions with protein targets.
Although fragments tend to have lower affinities for their targets, they exhibit high “binding efficiency per atom,” making them ideal for designing highly potent and selective drug candidates. By integrating fragment screening with structural biology and medicinal chemistry, FBDD accelerates the path from hit discovery to lead optimization, reducing costs, improving success rates, and broadening the reach of modern drug discovery.
FBDD Workflow: A Step-by-Step Approach
-
Fragment Library Design
The FBDD process begins with the careful selection or design of a diverse library of small molecule fragments. These fragments typically have low molecular weight (<300 Da) and high solubility, making them suitable starting points for drug discovery. Fragment libraries can be assembled from commercially available compounds, natural product extracts, or computationally designed molecules.
-
Fragment Screening
Fragments are screened against the target protein using a variety of biophysical techniques, including nuclear magnetic resonance (NMR) spectroscopy, X-ray crystallography, surface plasmon resonance (SPR), or isothermal titration calorimetry (ITC). These techniques enable researchers to detect weak binding interactions between fragments and target proteins.
-
Hit Identification and Validation:
Promising Fragments that show binding affinity and ligand efficiency are confirmed using additional orthogonal test methods (biochemical and biophysical assays) to ensure they are genuine start points, termed hits. Hit compounds are evaluated further to ensure that they have the potential to modulate the desired biological pathway or target and offer scope to be elaborated and optimised.
-
Fragment Elaboration and Optimization
Hit fragments are elaborated and optimized through a combination of rational drug design and medicinal chemistry techniques. By iteratively modifying the chemical structure of the fragments, researchers aim to enhance binding affinity, selectivity, and pharmacokinetic properties to develop lead compounds with drug-like characteristics. Structural information obtained from fragment screening facilitates rational drug design, guiding the optimization process.Fragments are chemically optimized by adding or modifying functional groups to enhance binding strength, specificity, and drug-like properties.
-
Lead Optimization
Lead compounds identified through FBDD undergo further optimization to improve their drug-like properties, including potency, selectivity, solubility, and metabolic stability. This iterative process involves medicinal and computational chemistry, structural biology, broader biological and safety evaluation involving a range of preclinical studies to develop candidates suitable for clinical testing. The lead optimization phase also involves exploration structure-activity relationships (SAR – making changes to molecules and observing the impact of profiles and properties), ADME profiling, and in-vivo efficacy studies to ensure pharmacokinetic and safety readiness for preclinical development.
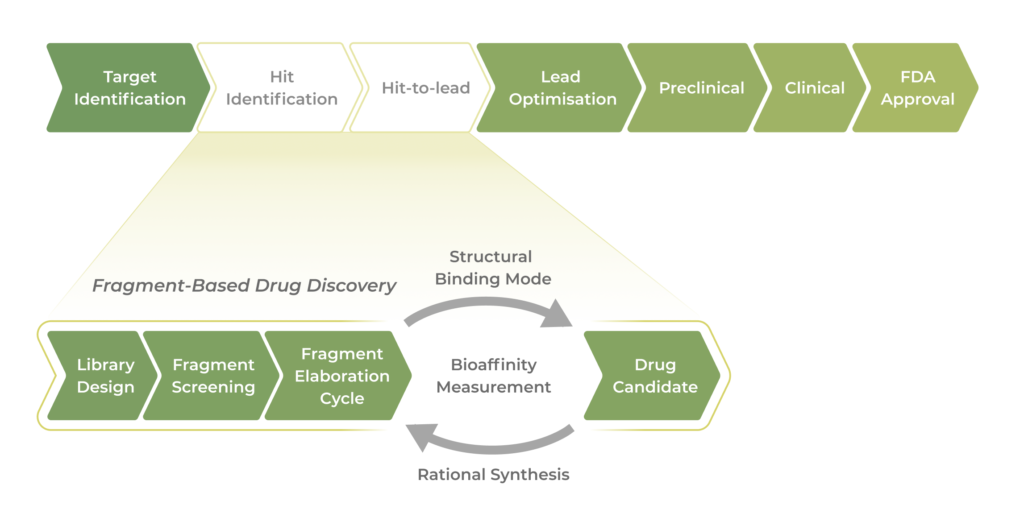
Why is FBDD a Preferred Approach?
-
Efficient Use of Resources:
FBDD focuses on screening smaller libraries of fragments, reducing the time and resources required for hit finding compared to traditional methods. Screening smaller sized molecules with more complimentary interactions with the target protein allows researchers to more efficiently explore a larger chemical space and increases the likelihood of identifying robust hits to rapidly explore their potential to generate high quality, novel lead compounds.
-
Higher Hit Rates:
As fragments are smaller than traditional drug sized molecules, they have a higher probability of binding to key sites on a protein (albeit with weak/modest affinity). This increases the likelihood of identifying a range of hits providing scope and opportunities that can be further optimized into potent drug candidates. This enhances the success rate of FBDD campaigns, generally identifying lead compounds with greater therapeutic potential.
- Improved Druggability: FBDD enables researchers to target challenging drug targets and protein-protein interactions that may be inaccessible using traditional methods. By focusing on smaller, more flexible molecules, FBDD can explore diverse chemical spaces and identify lead compounds with improved druggability.
- Rational Drug Design: Structural information when available to support fragment screening facilitates rational drug design, allowing researchers to rapidly optimize fragment hits through structure-based approaches (either modifying the fragment to increase affinity, or allowing understanding of the best places to expand and grow the fragment to pick up additional new interactions to improve potency/affinity.
These benefits make Fragment-based drug discovery (FBDD) a game-changer, especially for developing therapeutics for “hard-to-drug” targets.
How o2h Discovery Supports FBDD Programs?
At o2h discovery, our scientists specialize in delivering tailored Fragment-Based Drug Discovery services, helping to identify, validate, and optimize fragments into high-quality leads. Our expert medicinal chemists, in collaboration with leading academic computational chemists, have meticulously designed a fragment library that ensures pharmacophore diversity, molecular complexity, and optimal physicochemical characteristics tailored for Fragment-Based Drug Discovery (FBDD). Our library is continually expanded to address any gaps, ensuring comprehensive coverage for FBDD applications, with >50% of the library being synthesised in house.
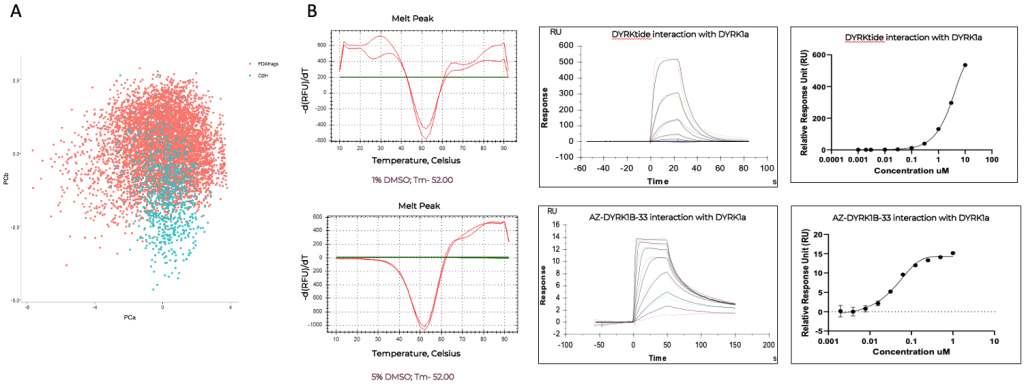
We leverage Biacore T200 Surface Plasmon Resonance (SPR) as a robust primary screening platform, enabling efficient evaluation of fragment binding at high micromolar concentrations. Fragments demonstrating confirmed activity are validated through orthogonal methods such as thermal stability (TS) assays.
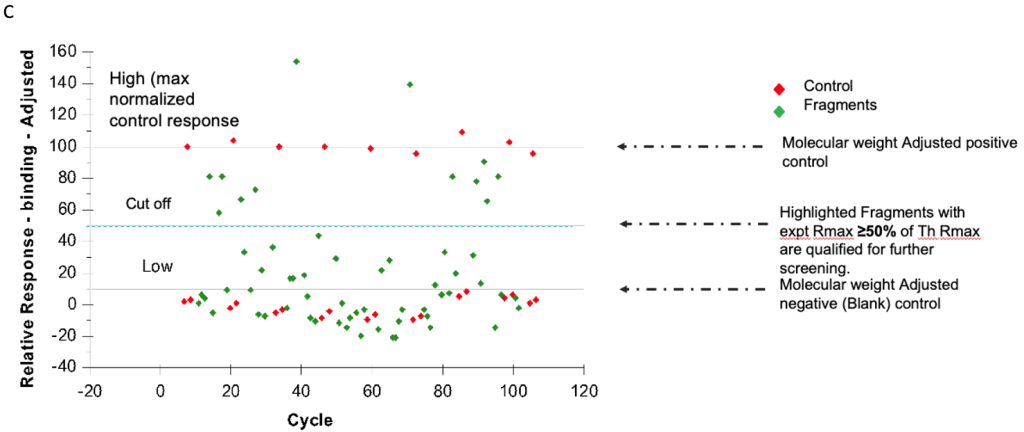
Fragment-based drug discovery development strategy
Our SPR assay development team employs a precise experimental approach, including:
-
- Target immobilisation based on research precedents.
- Buffer scouting and binding studies using natural peptides or control compounds.
- Competitive and allosteric interaction studies.
- Sensorgram analysis for Kd, Kon, and Koff with optimized signal response.
- Protein stability and responsiveness evaluation.
Fragment screening identifies hits with >50% theoretical Rmax activity, followed by detailed kinetic characterization through:
-
- Specific vs. non-specific interaction discrimination.
- Counter screens using mutant proteins or controls.
- LC-MS validation for identity and purity.
We support orthogonal validation (e.g., fluorescence-based thermal stability) and evolve shortlisted fragments via:
-
- “SAR by catalogue” and analogue synthesis.
- Structural and computational insights for binding mode analysis.
- Mutagenesis studies to enhance SAR and lead optimization.
Case Study: DYRK1A – From Fragments to Functional Leads
o2h discovery utilises Biacore T200 SPR to screen a diverse library of fragments against a challenging target. By analyzing binding kinetics and affinities, we identified fragments with high binding efficiency. These fragments underwent further optimization, resulting in a lead compound with significantly enhanced potency and drug-like properties. The project demonstrated the power of integrating FBDD with cutting-edge technology and expertise.
Dual-specificity tyrosine-regulated kinase 1A, DYRK1a Kinase target – Catalytic domain
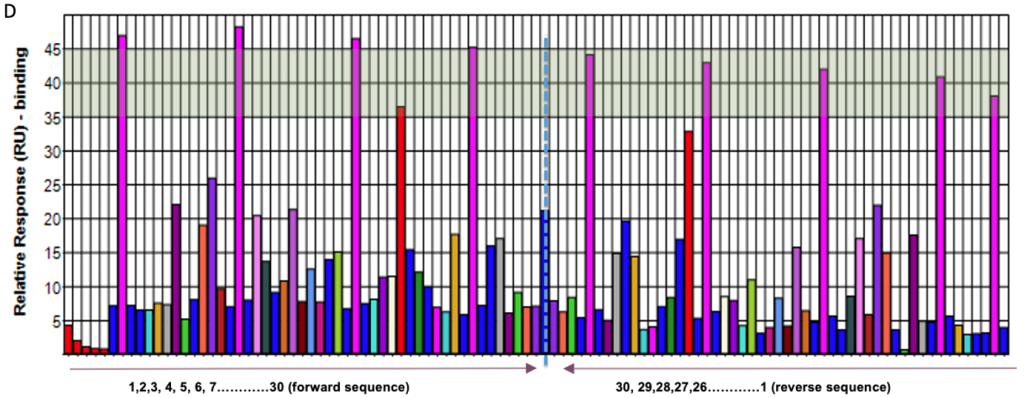
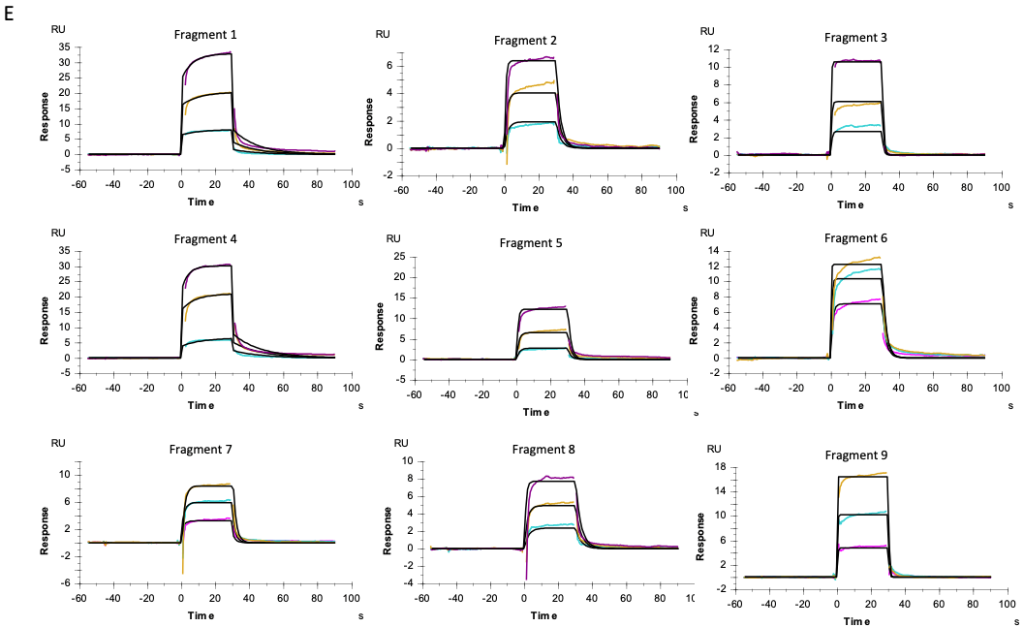
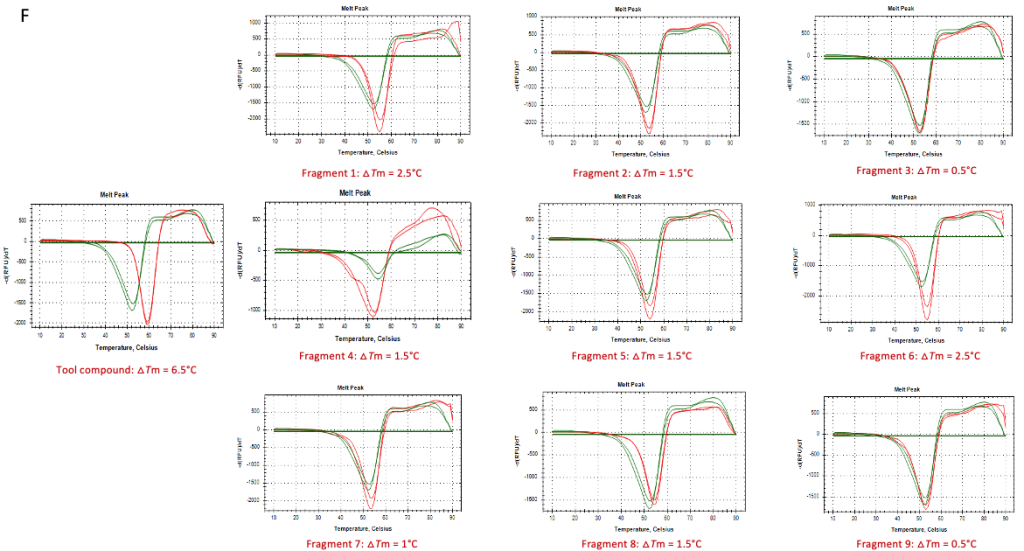
The Future of FBDD
Fragment-Based Drug Discovery continues to evolve as the pharmaceutical industry embraces artificial intelligence (AI) and advanced screening technologies. Modern workflows now combine biophysical screening tools like Surface Plasmon Resonance (SPR) with machine learning models, enabling deeper insights into fragment binding and accelerated generation of lead compounds.
For instance, a 2024 study in Nature Communications Chemistry highlighted how molecular fragmentation techniques are becoming key tools in AI-driven drug design, helping researchers break down complex molecules into simpler, more useful fragments.
Similarly, recent work introducing DigFrag, a digital fragmentation algorithm based on graph deep learning, has shown how AI can generate new fragment structures with greater diversity expanding the chemical space explored in FBDD and improving the chances of identifying high-quality hits.
These advances make FBDD particularly well suited for addressing hard-to-drug targets and unmet medical needs, including protein-protein interactions, epigenetic regulators, and novel therapeutic modalities.
Partner with o2h discovery
With our expertise in Fragment-Based Drug Discovery (FBDD) and access to cutting-edge tools like Biacore T200 SPR, we can help accelerate your research, by offering strategic planning and flawless execution to transform innovative pharmaceutical ideas into reality. Reach out to us at discovery@o2h.com to get a quote.



